physique 3 h
|
|
|
|
|
|
Un sachet de poudre blanche est saisi par les services de police et amené au laboratoire. Il s'agit d'analyser la composition de cette poudre. L'examen sous loupe binoculaire révèle que la poudre est composée de deux types de grains blancs séparables par leur granulométrie. On note F les grains fins et G les grains les plus gros. Des tests de solubilité sont alors entrepris :
- Dans l'eau, G est soluble, F est insoluble.
- Dans l'alcool absolu G est insoluble, F est insoluble.
- Exposer brièvement les méthodes de
séparation que vous connaissez.
- Laquelle semble la plus appropriée à notre cas. Justifier. - On désire identifier les deux composants
constituant la poudre. Etude des
grains de G :
a - On enregistre le spectre infrarouge par FTIR des grains. Donner le nom des principaux constituants d'un spectromètre infrarouge, légende de A à E, représentés sur le schéma ci-dessous.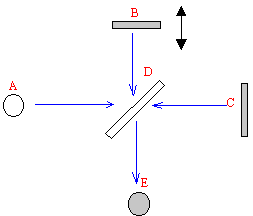
Où placeriez vous l'échantillon ? ( répondre comme suit : entre X et Y)
b - Le spectre de l'échantillon est représenté ci-dessous :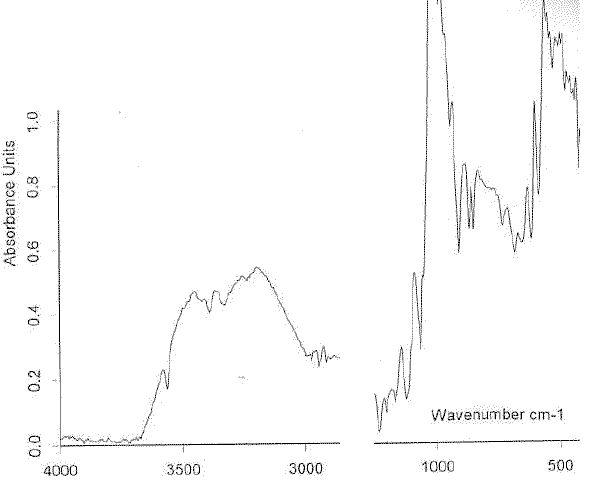
- Quelle réflexion vous inspire la qualité de celui-ci ? Comment pourrait-on l'améliorer ?
- De quel type de pollution atmosphérique le plus courant doit-on s'affranchir en spectroscopie IR ?
- Si les modes de vibrations sont inactifs en IR connaissez vous une autre méthode spectroscopique qui permet de les identifier ?
- Les modes de vibration de la molécule de CO2 ( ci-dessous ) sont -ils actifs en IR ?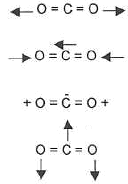
- Donner les zones spectrales ( en cm-1) qui sont précisées dans le tableau ci-dessous :
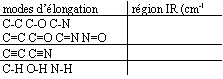
- Il s'avère que les grains G sont des grains de sucre. Proposez une mise en évidence colorimètrique simple des sucres.
- D'autre part les grains F s'avèrent être de la calcite, dont l'un des composants est le calcium - Etude des grains F. Termes
spectraux et structure fine de l'atome de calcium
20Ca.
- A quelle famille chimique appartient le calcium ? Est-il plus facilement oxydable ou réductible ? Justifier. - Un électron de la dernière couche de
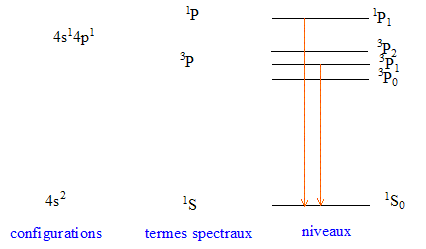
l'atome de calcium est excité vers un niveau 4p.
Donner la configuration électronique de cet
état excité et les termes spectraux
correspondant à celle-ci.
- Préciser la structure fine de chaque niveau en tenant compte du couplage spin-orbite
- Représenté schématiquement ces résultat dans un diagramme d'énergie. ( niveaux 4s et 4p)
- Parmi les 3 spectres de fluorescence X, lequel correspond au spectre du Ca ?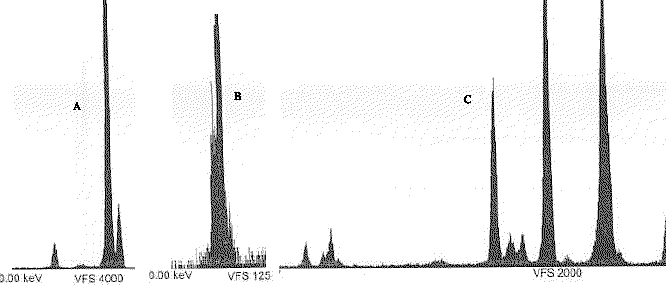
corrigé
méthodes de séparation :
liquide liquide :décantation de liquide non miscibles, phénomène de partage entre deux solvants
liquide gaz : distillation, entraînement à la vapeur d'eau
solide liquide : filtration, solubilité d'un solide dans un solvant, échangeurs d'ions, tamis moléculaires, échange entre un précipité et une solution, fusion et solidification fractionnées.
solide gaz : adsorption d'un gaz par un solide.
G et F peuvent être séparés en utilisant leur différence de solubilité dans l'eau ( seul G est soluble dans l'eau)
spectre infrarouge FTIR :
La Spectroscopie Infrarouge à Transformée de Fourier (FTIR) est basée sur l'absorption d'un rayonnement infrarouge par le matériau analysé. Elle permet via la détection des vibrations caractéristiques des liaisons chimiques entre deux atomes d'effectuer l'analyse des fonctions chimiques.
La spectrométrie IRTF est basée sur l'obtention dans le domaine temporel d'un interférogramme à l'aide d'un interféromètre de Michelson. Le spectre désiré est ensuite obtenu par transformée de Fourier de cet interférogramme.
L'interféromètre de Michelson est constitué de deux miroirs B et C, d'une lame séparatrice semi-transparente D;
A est la source lumineuse et E le plan d'observation ( détecteur)
L'échantillon doit être placé entre l'un des miroirs et la lame D.
Le spectre proposé présente une bande large dans la zone 3000 - 3500 cm-1 ( liaison hydrogène): en travaillant avec un solvant apolaire, on obsevera dans cette zone des pics interpétables.
Le spectromètre doit être entièrement purgé à l'air sec de façon à minimiser les absorptions dues à la vapeur d'eau et au dioxyde de carbone.
Seules les vibrations qui font varier le moment dipolaire de la molécules absorbent les radiations infrarouges. Parmi les modes proposés le premier et le 3ème modes de vibration sont inactifs.
Si des modes de vibrations sont inactifs en IR une autre méthode spectroscopique permet de les identifier : Raman
C-C (alcanes) : 600 à 1500 cm-1 faible ; C-O ( alcool, ether ): 1000 à 1300 cm-1 forte.
C-N ( amine aromatique) absorption forte à 1300 cm-1.
C=C (alcènes) : 1600 à 1680 cm-1 variable ; C=O (aldehyde, cétone, ester) : 1700 à 1750 cm-1 forte ;
triple liaison carbone carbone ou carbone azote (alcynes, nitrile) : 2100à 2250 cm-1 variable ;
C-H ( alcanes ) 2850 à 2960 cm-1 forte ; N-H (amine) 3300 à 3500 m-1 ;
O-H ( alcool ) 3200 à 3400 cm-1 forte , large ( associassion par liaison hydrogène);
test à la liqueur de Fehling qui est un réactif permettant la mise en évidence de sucres réducteurs .
Le calcium
appartient à la famille des alcalino-terreux, métaux facilement oxydables en ion M2+ ( perte aisée de deux électrons)
Ca ( Z = 20) : 1s2 2s2 2p6 3s2 3p6 4s2 dans l'état fondamental
1s2 2s2 2p6 3s2 3p6 4s1 4p1 dans un état excité
Le mouvement orbital de l'électron donne naissance à un champ magnétique interne, champ qui peut interagir avec l'aimantation (moment magnétique) intrinsèque associée au spin du même électron. On appelle cette interaction (faible), couplage spin-orbite.
Dans l'état fondamental 4s2 :
L'un des électrons a pour nombre quantique : l1=0 et s1 = ½ ; l'autre a pour nombre quantique : l2=0 et s2 = -½ ;
Composition du moment cinétique total : |l1-l2|<= L<= l1+l2 ; d'où L = 0
Composition du spin total : |s1-s2|<= S<= s1+s2 ; d'où S = 0
Il y a un seul couple (L,S) : L=S=0 non dégénéré ( L+1)(2S+1) = 1
couplage spin orbite : |L+S|<=J <= L+S ; d'où J=0
Le terme spectral correspondant à l'état fondamental 4s2 est : 1S0.
Dans l'état excité 4s1 4p1
L'un des électrons a pour nombre quantique : l1=0 et s1 = ½ ; l'autre a pour nombre quantique : l2=1 et s2 =½ ;
Composition du moment cinétique total : |l1-l2|<= L<= l1+l2 ; d'où L = 1
Composition du spin total : |s1-s2|<= S<= s1+s2 ; d'où S = 0 ; 1
Il y a deux couples (L,S) : L=1 ; S=0 et L=1 et S=1
couplage spin orbite : L=1 ; S=0 donne J= 1 ; L=1 et S=1 donne J= 2, 1, 0
Les termes spectraux correspondants à l'état excitél 4s1 4p1 sont : 2S+1PJ soit 1P1 (L=1; S=0; J=1 )
3P0 (L=1; S=1; J=0 ) ; 3P1 (L=1; S=1; J=1 ) ; 3P2 (L=1; S=1; J=2 ).
Seules certaines transitions sont permises ; les règles de sélection sont : DL=+1 ou -1 ; DJ=+1 ou -1 ou 0 ( mais la transition J=0 vers J=0 n'est pas possible)
La fluorescence correspond à l'émission de lumière visible par une substance sous l'excitation d'une radiation visible ou invisible d'onde plus courte. Lorsqu'un rayon lumineux frappe un corps, les électrons de celui-ci sont excités. Lorsqu'ils reviennent à leur état " normal ", ils émettent une énergie électromagnétique, ce qui produit la fluorescence.
si on excite l'atome par une source d'énergie, par exemple une lumière ultraviolette, les électrons sautent sur des orbites supérieures en absorbant l'énergie du rayonnement UV. Cet état est temporaire et les électrons ont tendance à reprendre leur position d'origine en restituant le surplus d'énergie absorbée sous la forme de lumière. Cette lumière est normalement dans une longueur d'onde plus longue que la lumière UV qui a permis d'exciter les électrons et se situe généralement dans le spectre visible. Cette lumière est appelée fluorescence.
Si les électrons mettent un certain temps à retour - menuner à leur état normal, quand la source de lumière ultraviolette est coupée, et que le minéral reste encore lumineux, alors ce minéral à la propriété de phosphorescence.
- Domaine
électromagnétique :
- Ordonnancez sur un axe graduer en longueur d'onde décroissante les domaines électromagnétiques suivants : infrarouge, microonde, ondes hertzienne, rayons X, rayons cosmiques, visible, audio-fréquences, ultraviolet et rayons g.
On ne demande pas les valeurs des bornes de chacun des domaines, seulement un classement par longueur d'onde décroissante
- Quelle est la condition préalable à toute étude par spectrométrie de masse et RMN ? En est-il de même pour la spectrométrie UV ? Laquelle des spectrométries précédemment citées nécessite l'application d'un champ magnétique extérieur ? - Chromatographie :
- Que vous inspire le chromatogramme ci-dessous ? Qu'envisagez vous de faire pour l'amméliorer ?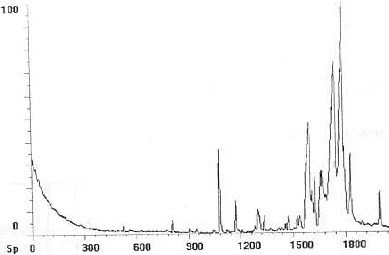
- Avec quel type de spectrométrie associe t-on généralement la HPLC ?
- Spectrométrie de masse :
- Quelles sont les conditions thermiques de stabilité du produit et la masse molaire maximale qui permettent une étude par la technique deCG-MS ? Si le produit n'est pas stable thermiquement, quelle autre méthode chromatographique peut-on employer ?
- Comment peut-on identifier assez facilement sur un spectre de masse la présence d'hydrocarbures linéaires saturés, aromatiques, de composés chlorés, bromés ou soufrés ou d'un aldehyde ? Peut-on identifier un composé contenant de l'iode par le même raisonnement que celui qui permet d'identifier les composés chlorés, soufrés ou bromés ?
- Si en spectrophotométrie de masse par impact électronique, le pic moléculaire d'un produit est absent ou incertain( donnez des exemples si vous en connaissez ) comment peut-on procéder pour les repérer tout de même ? Expliquer pourquoi ce pic moléculaire est absent ?
- Peut-on faire de la spectrométrie de masse directement sur des solides ? Donner des exemples.
- Quels éléments chimiques aisément dentifiables contiennent les corps dont on représente les pics remarquables si-dessous ?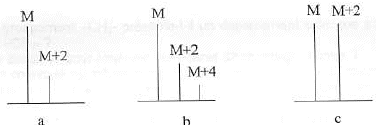
- Spectrométrie UV :
- Est-ce que l'eau et le méthanol peuvent servir de solvants dans l'UV proche (400-200 nm) ? Justifier.
- Quels sont les phénomènes dénommés A etB sur le schéma suivant ?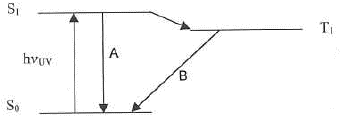
- RMN : expliquer le spectre ci-dessous en
répondant aux questions suivantes.
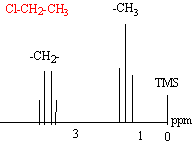
- Pourquoi le groupe CH2 présente-t-il un déplacement chimique supérieur à celui du groupe CH3 ?
- Quelle est la condition pour observer des figures de couplage simples ?
- Est- ce que le couplage est influencé par le solvant et l'intensité du champ magnétique B0 ?
- Comment fait-on pour prévoir le nombre de bandes d'un proton couplé avec un autre proton, ainsi que leurs intensités ?
- Quel phénomène se produit entre les protons liés à des hétéroatomes et le solvant ?
- Que se passe-t-il lorsque les protons sont sur des carbones liés à des atomes comme le chlore, le brome ou l'iode ? Et à des atomes comme 19F et 31P ? - Microscopie électronique à balayage
(MEB) :
- Sous l'action d'un faisceau d'électrons incident d'énergie Ep, deux types d'interactions entre les particules corpusculaires et la matière vont avoir lieu. Expliquer-les puis complétez le schéma ci-dessous :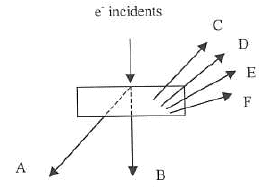
- Quelles sont les contraintes que doivent supporter un échantillon placé et analysé dans un microscope électronique à balayage ?
- Qu'est-ce qu'une métallisation ? A quoi sert-elle ? Comment est-elle réalisée et avec quels types de matériaux ?
corrigé (partiel)
domaines électromagnétiques :
par ordre de longueur d'onde décroissante :
ondes radio ; ondes hertziennes ; micro-ondes IR; visible ; UV ; rayons X ; rayon g ; rayons cosmiques
la condition préalable à toute étude par spectrométrie de masse et RMN est la présence de noyaux H et C.
Il n'en est pas de même pour la spectrométrie UV.
Spectroscopie de masse :
Le spectrographe de masse consiste à ioniser par des électrons une molécule A. Celle-ci va donc donner une entité A+ ayant perdu un électron. A+ va pouvoir se scinder en plusieurs groupements (chargé + ou non) plus petits, ou bien se réarranger.
On accélère alors ces particules par un champ électrique, puis elles sont déviées par un champ magnétique. On montre que la déviation est proportionnelle à m /q (ici à m/e).
On introduit des ions positifs entre deux plaques à différent potentiel : il s'ensuit une accélération des ions. Leur énergie cinétique est alors: Ec = e.V (avec e la charge de l'ion et V la différence de potentiel entre les deux plaques).
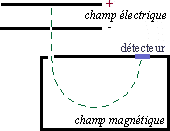
Arrivé dans un champ magnétique, l'ion subit une force F = H.e.v = (m.v²)/R (avec H la valeur du champ magnétique, m la masse de l'ion, v sa vitesse et R le rayon de la trajectoire décrite par l'ion dans le champ magnétique). Alors, on a la relation: (e/m) = (2.V)/(H².R²)
Il est donc facile de sélectionner une certaine valeur de (e/m) en faisant varier V ou H.
Ce dispositif très sensible permet de connaître tous les différents isotropes présents dans le matériau à tester, ainsi que leur quantité. Mais pour cela il faut introduire des ions gazeux. De plus un vide poussé est nécessaire pour cette spectroscopie. Cette technique est donc lourde d'utilisation
La spectroscopie RMN utilise un champ magnétique extérieur.
chromatographie liquide ou HPLC :
Elle peut ne comporter qu'un appareillage simple, constitué d'une colonne de verre remplie de particules de phase stationnaire, le solvant migrant sous le simple effet de la pesanteur. Toutefois, dans de telles conditions, la résolution obtenue est relativement médiocre.
Les progrès sont venus de la possibilité de fabriquer des particules de très faible taille (3 - 10 µm) et de granulométrie homogène, qui permettait de réaliser des colonnes très efficaces, mais au prix d'une augmentation considérable de la résistance au passage du solvant (= perte de charge), impliquant la nécessité d'employer des pompes à haute pression pour pousser les solvants (et des tubes résistants pour y mettre les phases stationnaires) : ainsi naquit la chromatographie liquide à haute performance/pression (C.L.H.P. - ou HPLC en anglais).
Une bonne séparation se traduira par une séparation distincte des pics correspondant à chacun des produits.
Le chromatogramme proposé n'est pas très lisible dans la zone 1500 - 1800 : pour l'améliorer on peut changer de solvant. (en modifiant la polarité de la phase mobile )
détecteur UV-visible :
il mesure l'absorption de la lumière par le produit à la sortie de la colonne. E opère à longueur d'onde constante, celle-ci ayant été fixée par l'opérateur. La lampe Deuterium est utilisé pour des longueurs d'ondes variant de 190-350 nm et la lampe à vapeur de mercure est utilisé à la longueur d'onde non variable de 254 nm. Pour que ce type de détecteur soit utilisable, il faut que :- le produit à détecter absorbe la lumière à une longueur d'onde accessible à l'appareil, et que son coefficient d'absorption e soit suffisamment grand ;
- la phase mobile n'absorbe pas la lumière à la longueur d'onde choisie par l'opérateur.
On associe généralement la HPLC avec un spectrographe de masse CG-MS.
spectroscopie de masse :
Pour les produits les plus stables thermiquement ( Teb <250°C ) , la masse molaire peut atteindre 1000 g/mol.
Si le produit n'est pas stable thermiquement, autre méthode chromatographique peut être employée : LC-MS
Les alcanes donnent surtout des cations éthyl, propyl, butyl et pentyl, quel que soit l’alcane étudié.
dérivés aromatiques : présence de pic correpondant au noyau benzénique
Dérivés carbonylés : il y a également rupture en a de la fonction carbonyle ; les aldéhydes peuvent perdre H , et le spectre de masse présentera un pic M – 1.
On trouve dans ce spectre un pic important : le pic de base, et on mesure les autres pics en % par rapport au pic de base. On retrouve aussi des pics de masse molaire M+1 et M+2 provenant des isotopes du carbone, de l’azote, de l’oxygène,....
les composés chlorés, bromés, soufrés présentent des amas isotopiques
soufre : 32S( abondance relative 100%) 34S (abondance relative 4,4%)
35Cl ( 75%) et 37Cl ( 25%) ; ( schéma a) ( 100% pic M et 33 %, pic à M+2 )
2 atomes de chlore : ( schéma b)
brome : isotopes naturels (en %), 79Br (50,69), 81Br (49,31). ... ( schéma c)
le pic moléculaire peut être absent si l'un des fragments de la molécule est plus stable. En faisant la somme des masses des deux fragments les plus abondants, on retrouve le pic moléculaire.
exemple : CH3-COO-CH2-CH=CH2 ; pic le plus abondant CH3-CO+ m/e = 43
On peut faire de la spectrométrie de masse directement sur des solides : par Maldi-TOF
UV :
la spectroscopie UV-visible s'occupe des électrons de valence. Les transitions possibles seront donc entre les électrons des orbitales moléculaires (OM) liantes ou non liantes et orbitales moléculaires anti-liantes.
Les trois types d'OM rencontrées dans les molécules organiques (en absence de métaux de transition) sont les OM non liantes (n), les OM liantes ou anti liantes s et p (respectivement s * et p *).
La molécule dont on réalise le spectre est susceptible d'interagir avec le solvant. Il s'agit généralement de stabilisation par liaison hydrogène. Qui dit stabilisation dit énergie de transition plus faible, donc longueur d'onde plus grande. Des molécules possédant des atomes susceptibles d’interagir par liaison hydrogène verront la longueur d'onde d'absorption augmenter si le solvant est de plus en plus polaire.
Le même type de raisonnement peut-être étendu à des considérations sur les transitions p* -->p* et n -->p*. On en déduit une stabilisation dans les solvants polaires comme l'eau et le méthanol.
phénomène A : l'atome passe d'un niveau excité à un niveau de moindre énergie :
l'énergie libérée est emportée par un photon émis ( transition radiative)
phénomène B : transition non radiative
Le processus non radiatif permet d'éliminer l'énergie accumulé par transfert vers d'autre molécules ou par vibration-rotation (émission IR) jusqu'au niveau de vibration le plus bas et le niveau électronique fondamental
RMN du chlorure d'éthyle :Cl-CH2-CH3.
Les hydrogènes du groupe CH2 et du groupe CH3 présentent des environnements magnétiques différents.
Du fait de la présence de l'atome de chlore proche ( atome attracteur) , le déplacement chimique des H du groupe CH2 est plus important que le déplacement des H du groupe CH3.
La taille du pic est proportionnelle au nombre d'hydrogène qu'il représente.
Bien souvent on observe des multiplets : un système AmXp admet (p+1) pics pour les protons HA et ( m+1) pics pour les protons HX. Les tailles des pics ont les coefficients du binôme de Newton. L'écartement des pics est égal à la constante de couplage J : elle est indépendante du champ magnétique appliqué.
Les protons sont sur des carbones liés à des atomes comme le chlore, le brome ou l'iode : déplacement chimique du signal, pas de couplage spin spin entre H et l'hétéroatome. Par contre les noyaux 19F et 31P ont des propriétés magnétiques, donc couplage spin spin..
expérimentalement, on constate que, placés dans un champ magnétique de l’ordre de 1,4 T (Tesla), les protons absorbent l’énergie électromagnétique vers 60.106 Hz (fréquence radio). Placés dans le même champ, les noyaux du carbone 13 (13C) absorbent vers 15.106 Hz. Les noyaux de fluor 19 (19F) absorbent vers 56.106 Hz
le couplage spin spin est influencé par le solvant.
phénomène se produisant entre les protons liés à des hétéroatomes et le solvant : liaison H
MEB :
Un MEB utilise un faisceau d'électrons à la place des photons utilisés dans un microscope optique.
Ceci permet de résoudre les deux inconvénients de la source lumineuse : la longueur d'onde du faisceau électronique est 100000 fois plus faible que celle de la lumière, et d'autre part l'ouverture de ce faisceau est très faible.
Le faisceau en touchant la surface de l'échantillon produit les interactions suivantes:
- des électrons secondaires : sont émis lorsque le faisceau primaire qui a perdu une partie de son énergie excite les atomes de l'échantillon. Les électrons secondaires possèdent une énergie faible (autour de 50 eV) suivant un large spectre.
- des électrons rétrodiffusés : électrons du faisceau primaire qui ont réagi de façon quasi élastique avec les atomes de l'échantillon. Ils sont renvoyés dans une direction proche de leur direction d'origine avec une faible perte d'énergie.
- des rayons X : radiations électromagnétiques. L'énergie des photons X émis dans le MEB est comprise entre 0.5 et 30 KeV. Le faisceau d'électrons du microscope est capable d'éjecter des électrons des différentes couches électroniques des atomes constituant le matériau observé. Lorsqu'un électron est éjecté il est remplacé par un électron d'une couche supérieure.
Si le faisceau est capable d'éjecter des électrons de la couche la plus profonde il y aura donc émission de toutes les raies caractéristiques de l'atome c'est le spectre de raies.
La métallisation consiste à recouvrir de métal des objets non métalliques (cuir, plastiques, résine, platre, bois etc.) dans un but de décoration, de protection, de durcissement ou de régénération.
La métallisation sous vide consiste à placer dans une enceinte à vide la pièce à métalliser, en présence d'une masse de métal chauffé jusqu'à sa température de vaporisation et dont les vapeurs viennent se déposer en couches minces et régulières sur tous les recoins de la pièce.
La métallisation au pistolet, dont le résultat s'apparente à celui de la galvanisation, mais qui s'applique à des pièces de grandes dimensions, consiste à pulvériser du métal fondu à bas point de fusion au moyen de pistolets spéciaux.
par électrolyse dans le cas d'objet métallique : pièce placée à la cathode négative ; solution contenant un sel du métal à déposer.
contraintes que doivent supporter un échantillon placé et analysé dans un microscope électronique à balayage : vide poussé